Satan/Loree McBride Using Genetic Modification Interdimensionally on Human Race
First off, I’d like to state that my gastrointestinal problems appear to be an inability to digest fructans (specfically lack of ability to produce the enzyme Alpha-galactosidase) and an inability to digest lactose, which seem connected to a genetic disorder. Doing research on what causes my body’s inability to produce alpha-galactosidase, it seems to me that I have a rare genetic disorder that generally affects males worse than females, called Fabry disease. It appears I also have lactose intolerance, which appears to be a separate genetic disorder.
Satan seems to like to mess with our genes and I suspect that the reason the coronavirus vaccine makes people retarded may be using some sort of interdimensional science to modify the genes in the brain, bringing on retardation, which is accelerated by something in the coronavirus vaccine, enabling Satan and Loree to bring on mental retardation using genetic manipulation to the brains of those who’ve had the coronavirus shot.
If you want to laugh this off, remember that my IQ is higher than Satan’s right now! This means I am a mega-genius. My symptoms seem to defy most illness categories and I think I have a mild case of Fabry disease, which Satan is attempting to accelerate now, perhaps modifying my genes using interdimensional science. I’m not worried about it, because my husband is the most brilliant doctor in the world and I don’t plan to visit any doctors right now, since I think most of them are retarded from getting the coronavirus vaccine! I have faith in Jesus. I bet those Gail Commandments are designed to help people with Fabry disease!
What is Fabry disease?
People who have Fabry disease don’t produce enough healthy versions of an enzyme (blood chemical) called alpha-galactosidase A (alpha-GAL). These enzymes prevent sphingolipids, a fat-like substance, from collecting in blood vessels and tissue.
Without functioning alpha-GAL enzymes, harmful levels of sphingolipids build up in blood vessels and tissues. Fabry disease affects the heart, kidneys, brain, central nervous system and skin. It is an inherited condition passed from parent to child. It’s sometimes called Anderson-Fabry disease.
What are the types of Fabry disease?
The types of Fabry disease reflect a person’s age when symptoms first appear. Types include:
- Classic type: Symptoms of classic Fabry disease appear during childhood or the teenage years. One hallmark disease symptom — a painful burning sensation in the hands and feet — may be noticeable as early as age two. Symptoms get progressively worse over time. NOTE: I have had burning sensations in my hands and feet for years, which I’ve pretty much overlooked. It started around my 40s. I always blamed it on a systemic yeast infection.
- Late-onset/atypical type: People with late-onset Fabry disease don’t have symptoms until they’re in their 30s or older. The first indication of a problem may be kidney failure or heart disease.
How common is Fabry disease?
Approximately one out of every 40,000 males has classic Fabry disease. Late-onset or atypical Fabry disease is more common. It affects about one in every 1,500 to 4,000 males.
Experts aren’t sure how many females have Fabry disease. Some females don’t have symptoms or have mild, easy-to-dismiss symptoms, so the condition frequently goes undiagnosed in women. NOTE: Jesus once told me I have masculine genes. I’m certain if I had not figured this out on my own, I probably would have been undiagnosed my entire life, especially since it’s pretty rare and affects males more than females.
SYMPTOMS AND CAUSES
What causes Fabry disease?
Children inherit a mutation (change) in the galactosidase alpha (GLA) gene on the X chromosome from a parent. The GLA gene produces the alpha-GAL enzyme that helps break down fatty substances (sphingolipids). People who inherit a defective GLA gene don’t produce enough alpha-GAL enzyme. As a result, fatty substances build up in blood vessels.
Who might get Fabry disease?
People with Fabry disease inherit a mutated gene on the X chromosome from a parent. Males inherit one X chromosome from their mothers. Females have two X chromosomes, one from each parent.
A parent can pass on the faulty gene that causes Fabry disease to a child in different ways:
- Fathers pass their X chromosome with the faulty gene to all of their daughters. All of these daughters will have the gene mutation that causes Fabry disease. Sons aren’t at risk because males get the Y chromosome from their fathers (not the X chromosome). NOTE: So it appears I got the faulty gene from my father OR Satan may have done it interdimensionally while I was in my mother’s womb, specifically attacking my King David genes. I almost died my first month of life from allergy to my formula and was very skinny as a kid, with “polio legs”.
- Mothers have a 50% chance of passing their affected X chromosome to their daughters or sons. Some family members can have the gene mutation while others don’t.
What are the symptoms of Fabry disease?
Symptoms of Fabry disease vary depending on the type. Some symptoms are mild and might not appear until later in life. Males tend to have more severe symptoms than females. NOTE: I appear to have a weak version of Fabry disease or some variation of it. I think Satan messed with my genes to try to kill me because I’m Jesus’ favorite. I’m not too worried about this, because my husband is a brilliant doctor. Fabry disease symptoms include:
- Numbness, tingling, burning or pain in the hands or feet. NOTE: I’ve had these symptoms for the past 20 years or more.
- Extreme pain during physical activity.
- Heat or cold intolerance. NOTE: This seems to have happened within the past 2 years. I don’t tolerate heat very well.
- Abnormal opacity of the eye (cornea), which does not change someone’s vision.
- Dizziness.
- Flu-like symptoms, including fatigue, fever and body aches.
- Gastrointestinal problems, such as diarrhea, constipation and abdominal pain. NOTE: This seems to have worsened considerably in the past 2 years.
- Hearing loss or ringing in ears (tinnitus).
- High levels of protein in urine (proteinuria). NOTE: I have had urinary frequency problems for at least 20 years (which can be caused by a high-protein diet OR high levels of protein in the urine). I always wrote this off as a yeast problem. But my yeast has been cured and I still deal with urinary frequency. I do recall that in the 1990s, they found a lesion on one of my kidneys that they said was harmless, but it could be a symptom of Fabry disease.
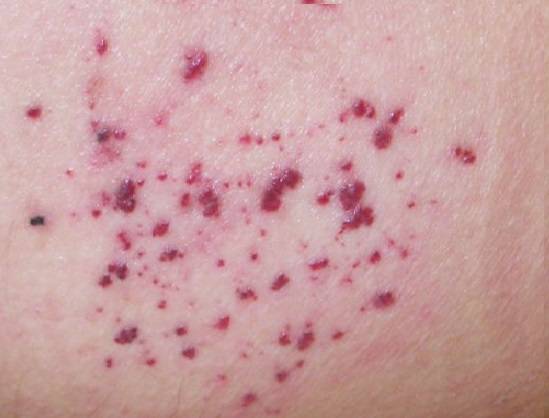
- Raised red or purplish skin lesions (angiokeratoma) on your chest, back and in the genital area. NOTE: See image below.
- Sweating less (hypohidrosis) or not at all (anhidrosis).
- Swelling (edema) in the legs, ankles or feet.
DIAGNOSIS AND TESTS
How is Fabry disease diagnosed?
Your healthcare provider may order tests to diagnose Fabry disease, including:
- Enzyme assay: This test measures alpha-GAL enzymes in blood. Measurements of 1% or lower indicate disease. This test is most reliable for males and should not be used in females.
- Genetic: Because females with Fabry disease can have normal levels of alpha-GAL enzymes, providers use genetic testing (DNA sequencing) to identify the GLA gene mutation. NOTE: Brent Spiner M.D. (my husband) could do this with the technology we have at Church of Gail. However, I suspect that Satan may be messing with our scanners using interdimensional warfare to cause them to be inaccurate and perhaps we need to take this into consideration.
- Newborn screenings: Some states test newborns for Fabry disease and other lysosomal storage disorders. The enzyme test is included as part of routine newborn screenings.
MANAGEMENT AND TREATMENT
How is Fabry disease managed or treated?
There isn’t a cure for Fabry disease. Medications for pain and stomach problems can ease symptoms. There are two treatments that may slow down the build up of the fatty substances with the goal to prevent heart problems, kidney disease and other life-threatening complications:
- Enzyme replacement therapy: Every two weeks, you receive an intravenous (IV) infusion of lab-made agalsidase beta enzyme (Fabrazyme®). This replacement enzyme does the work of the missing alpha-GAL enzyme so that fatty substances don’t build up. You may receive an antihistamine and other medications before therapy to prevent an allergic reaction.
- Oral chaperone therapy: Chaperones are small molecules that repair faulty alpha-GAL enzyme. The mended enzymes can then break down the fatty substance. With this therapy, you take a pill (migalastat [Galafold®]) every other day to stabilize the faulty alpha-GAL enzyme. Not everyone with Fabry disease can be treated with this medication. It depends on your specific genetic mutation in the GLA gene if you are eligible for this treatment.
Researchers are actively developing several new therapies using genetic engineering and stem cell technologies.
What are the complications of Fabry disease?
Years of build up of the fatty substance can damage blood vessels and lead to life-threatening problems, such as:
- Heart problems, including arrhythmia, heart attacks, enlarged heart and heart failure.
- Kidney failure.
- Nerve damage (peripheral neuropathy). NOTE: I suffer from peripheral neuropathy. I often get spasms in my toes and fingers, too.
- Strokes, including transient ischemic attacks (TIA or ministrokes). NOTE: My paternal grandmother died of ministrokes in her 80s and I appear to have gotten my bunions from her. It appears she had Fabry disease and I got it from her.
PREVENTION
How can I prevent Fabry disease?
Fabry disease is inherited. If you carry the mutated gene that causes Fabry disease, talk to a genetic counselor. This specialist can explain the chances of passing the gene to your children and discuss options. For instance, a process called preimplantation genetic diagnosis (PGD) identifies embryos that don’t carry the mutated gene. Your doctor implants healthy embryos during in vitro fertilization (IVF). PGD ensures your child won’t have the mutated gene or get Fabry disease.
OUTLOOK / PROGNOSIS
What is the prognosis (outlook) for people with Fabry disease?
Fabry disease is a progressive disease. Symptoms, and the risk of serious complications, worsen with age. People with Fabry disease have a higher risk of life-threatening problems that can shorten life expectancy. On average, males with classic Fabry disease tend to live to their late 50s. Females with the disease often live into their 70s. You may add years to your life by getting appropriate care for heart and kidney disease and taking steps to reduce strokes.
LIVING WITH
When should I call the doctor?
You should call your healthcare provider if you have Fabry disease and experience:
- Chest pain, irregular heartbeat, shortness of breath or signs of heart attack. NOTE: I often suffer from arrhythmias. This has been happening for at least 20 years.
- Excessive swelling or fluid retention. NOTE: When I stand up all the veins in my feet swell up and look grotesque. It’s been like this for years, at least since about 1987.
- Extreme dizziness, vision problems or signs of stroke.
- Hearing loss.
- Severe abdominal pain or diarrhea. NOTE: I have had intestinal issues for years. I suspect Satan has messed with my genes to cause me to have the lack of certain enzymes needed for proper digestion of certain sugar molecules especially lactose and fructans. Apparently, Satan messes with our scanners so that he can bring this on interdimensionally, so that the symptoms are inconsistent.
What questions should I ask my doctor? NOTE: I think most doctors are retards now from having gotten the coronavirus shot and only trust my doctor husband Brent Spiner M.D. I think my current strategy to deal with lactose intolerance and avoiding excessive intake of fructans (onions and garlic) will work for now.
You may want to ask your healthcare provider:
- How did I get Fabry disease?
- What type of Fabry disease do I have?
- What is the best treatment for me?
- What are the treatment risks and side effects?
- Are my family members at risk for developing Fabry disease? If so, should we get genetic testing?
- What type of ongoing care do I need?
- Should I look out for signs of complications?
Fabry disease is a serious genetic disorder that can lead to life-threatening heart and kidney problems. It’s a progressive disease that worsens over time. Symptoms may develop during childhood (classic type) or middle adulthood (atypical type). Males tend to have more severe symptoms. Some females have mild symptoms and don’t know they have the disease. If you have Fabry disease, talk to your healthcare provider about ways to lower stroke risk and protect your heart and kidneys. Newer therapies, including enzyme replacement and oral chaperone treatment, can help you manage the disease and reduce the odds of serious complications.
Understanding Fabry Disease
What is Fabry disease?
Fabry disease (FD) is a rare, inherited disease. It’s progressive and can be life-threatening. People with FD have a damaged gene that leads to a shortage of an essential enzyme. The shortage results in a buildup of specific proteins in the body’s cells, causing damage to the:
- heart
- lungs
- kidneys
- skin
- brain
- stomach
The disease affects both men and women in all ethnic groups, but men are usually more severely affected.
There are two types of FD. Type 1 FD, also known as classic FD, starts in childhood and is less common than type 2, which has a later onset. An estimated 1 out of 117,000 people has FD.
FD is named for Johannes Fabry, a doctor in Germany who first described its symptoms in 1898. It’s also known as Anderson-Fabry disease, for William Anderson, a British doctor who also noted it in that same year. Other names for FD are:
- galactosidase alpha (GLA) gene deficiency
- enzyme alpha-galactosidase A deficiency
- angiokeratoma corporis diffusum
- angiokeratoma diffuse
- ceramide trihexosidase deficiency
Fabry disease symptoms
FD has many different symptoms, making diagnosis difficult. Symptoms can vary between men and women, and between type 1 and type 2 FD.
Symptoms of type 1 FD
Early symptoms of type 1 FD include:
- Burning or tingling pain in the hands and feet. In males this can occur as early as 2 to 8 years old. In females it occurs later in childhood or adolescence. Episodes of intense pain, which can last from minutes to days, are called “Fabry crises.”
- Lack of sweat production. This affects more males than females.
- Skin rash. This reddish-purple rash is slightly raised and occurs between the belly button and the knees. It’s called angiokeratoma.
- Stomach problems. This includes cramps, gas, and diarrhea.
- Abnormal corneas. Blood vessels in the eyes may have a changed appearance, but this doesn’t affect vision.
- General tiredness, dizziness, headache, nausea, and heat intolerance. Males may have swelling in their feet and legs.
As type 1 FD progresses, symptoms become more serious. When people with type 1 reach their 30s and 40s, they can develop kidney disease, heart disease, and stroke.
Symptoms of type 2 FD
People with type 2 FD also develop problems in these areas, although usually later in life, in their 30s to 60s.
Serious FD symptoms vary from person to person and can include:
- A progressive decrease in kidney function, advancing to kidney failure.
- Heart enlargement, angina (heart-related chest pain), irregular heartbeat, thickening of the heart muscle, and eventually heart failure.
- Strokes, occurring in some men and women with FD in their 40s. This may be more common in women with FD.
- Stomach problems. About 50-60 percent of women with FD may have pain and diarrhea.
Other signs of FD include:
- hearing loss
- ringing in the ears
- lung disease
- intolerance of strenuous exercise
- fever
Pictures of Fabry disease

What causes Fabry disease?
Who inherits FD
A specific gene mutation causes FD. You inherit the damaged gene from your parents. The damaged gene is located on the X chromosome, one of the two chromosomes that determine your sex. Males have one X chromosome and one Y chromosome, and females have two X chromosomes.
A man with the FD gene mutation on the X chromosome will always pass it on to his daughters, but not to his sons. The sons get the Y chromosome, which doesn’t have the damaged gene.
A woman with the FD mutation on one X chromosome has a 50 percent chance of passing it on to her sons and daughters. If her son gets the X chromosome with the FD mutation, he will inherit FD.
Because a daughter has two X chromosomes, she may have less severe FD symptoms. This is because not all of her body’s cells will activate the X chromosome that carries the defect. Whether or not the damaged X chromosome is activated occurs early in your development and remains that way for the rest of your life.
How genetic mutations lead to FD
FD is caused by as many as 370 mutations in the GLA gene. Particular mutations tend to run in families.
The GLA gene controls the production of a particular enzyme called alpha-galactosidase A. This enzyme is responsible for breaking down a molecule in the cells known as globotriaosylceramide (GL-3).
When the GLA gene is damaged, the enzyme that breaks GL-3 down can’t function properly. As a result, GL-3 builds up in the body’s cells. Over time, this fatty buildup damages the cell walls of blood vessels in the:
- skin
- nervous system
- heart
- kidneys
The degree of damage FD causes depends on how severe the mutation in the GLA gene is. That’s why FD symptoms can vary from person to person.
How is Fabry disease diagnosed?
FD can be difficult to diagnose because the symptoms are similar to those of other diseases. Symptoms are often present long before a diagnosis. Many people are not diagnosed until they have an FD crisis.
Type 1 FD is most often diagnosed by doctors on the basis of the child’s symptoms. In adults, FD is often diagnosed when they’re being tested or treated for heart or kidney problems.
An FD diagnosis for males can be confirmed by a blood test that measures the amount of the damaged enzyme. For females, this test isn’t sufficient, because the damaged enzyme may seem normal even though some organs are damaged. A genetic test for the defective GLA gene is necessary to confirm whether a female has FD.
For families with a known history of FD, prenatal tests can be performed to determine if a baby has FD.
Early diagnosis is important. FD is a progressive disease, which means that symptoms get worse over time. Early treatment can help.
Fabry disease treatment options
FD can cause a wide variety of symptoms. If you have FD, you’ll probably see specialists for some of these symptoms. In general, treatment will aim at managing symptoms, relieving pain, and preventing further damage.
Once you’ve been diagnosed with FD, it’s important to regularly see your doctor to monitor your symptoms. People with FD are advised not to smoke.
Here are some FD treatment options:
Enzyme-replacement therapy (ERT)
ERT is now a first-line treatment recommended for all people with FD. Agalsidase beta (Fabrazyme) has been used since 2003, when it was approved by the U.S. Food and Drug Administration. It’s given intravenously, or through an IV.
Pain management
Pain management can involve avoiding activities that might bring on symptoms, such as strenuous exercise or temperature changes. Your doctor may also prescribe medications such as diphenylhydantoin (Dilantin) or carbamazapine (Tegretol). These are taken daily for pain reduction and prevention of FD crises.
For your kidney
A low-protein, low-sodium diet may help if you have a mildly reduced kidney function. If your kidney function gets worse, you may need kidney dialysis. In dialysis, a machine is used to filter your blood three times a week or more, depending on what type of dialysis you’re on and how much you need. A kidney transplant may also be necessary.
As-needed treatments
Heart problems will be treated as they are for people without FD. Your doctor may prescribe medications to manage the condition. Your doctor may also prescribe treatments to reduce the risk of stroke. For stomach problems, your doctor may prescribe medication or a special diet.
Complications of Fabry disease
One potential complication of FD is end-stage renal disease (ESRD). ESRD can be deadly if you aren’t treated with dialysis or a kidney transplant. Almost all males with FD develop ESRD. But only about 10 percent of females with FD develop ESRD.
For people who are treated to control ESRD, heart disease is a major cause of death.
Fabry disease outlook and life expectancy
FD can’t be cured, but it can be treated. Awareness of FD is increasing. ERT is a relatively new treatment that helps stabilize symptoms and lower the occurrence of FD crises. Research is ongoing for other treatment possibilities. Gene replacement therapy is in a clinical trial. Another approach in the research phase, called chaperone therapy, uses small molecules to stop the damaged enzyme.
Life expectancy for people with FD is lower than that of the general U.S. population. For males, it’s 58.2 years. For females, it’s 75.4 years.
A frequently overlooked FD complication is depression. It can be helpful to reach out to other people who understand. There are several organizations for people with FD which have resources that can help both people with FD and their families:
- Fabry Support and Information Group
- National Fabry Disease Foundation
- International Center for Fabry Disease
Alpha-galactosidase (α-GAL, also known as α-GAL A; E.C. 3.2.1.22) is a glycoside hydrolase enzyme that hydrolyses the terminal alpha-galactosyl moieties from glycolipids and glycoproteins. Glycosidase is an important class of enzyme catalyzing many catabolic processes, including cleaving glycoproteins and glycolipids, and polysaccharides. Specifically, α-GAL catalyzes the removal of the terminal α-galactose from oligosaccharides.[1]
The enzyme is encoded by the GLA gene.[2] Two recombinant forms of human alpha-galactosidase are called agalsidase alpha (INN) and agalsidase beta (INN). A mold-derived form is the primary ingredient in gas relief supplements.
Contents
- 1 Function
- 2 Reaction mechanism
- 3 Disease relevance
- 4 See also
- 5 References
- 6 Further reading
- 7 External links
Function
This enzyme is a homodimeric glycoprotein that hydrolyses the terminal alpha-galactosyl moieties from glycolipids and glycoproteins. It predominantly hydrolyzes ceramide trihexoside, and it can catalyze the hydrolysis of melibiose into galactose and glucose.α-GAL removing the terminal α-galactose
{kind=link}
Reaction mechanism
A double displacement reaction mechanism of α-GAL’s catalytic action.The ligand (black) when bound in the active site of the enzyme (blue). The two key amino acid residues in the active site are Asp-170 and Asp-231. First, Asp-170 performs a nucleophilic attack on the glycosidic bond to release the terminal α-galactose molecule from the ligand. Then, Asp-231 serves as an acid to remove a proton from water, making it more nucleophilic to attack the galactose-Asp complex and release α-galactose from the active site.[3][4][5]
{kind=link}
Disease relevance
Fabry disease
Signs and Symptoms
Defects in human α-GAL result in Fabry disease, a rare lysosomal storage disorder and sphingolipidosis that results from a failure to catabolize α-D-galactosyl glycolipid moieties.[6] Characteristic features include episodes of pain in hands and feet (acroparethesia), dark red spots on skin (angiokeratoma), decreased sweating (hypohidrosis), decreased vision (corneal opacity), gastrointestinal problems, hearing loss, tinnitus, etc.. Complications for this disease can be life-threatening and may include progressive kidney damage, heart attack, and stroke. This disease may have late onset and only affect the heart or kidneys.[7]
Fabry disease is an X-linked disease, affecting 1 in 40,000 males. However, unlike other X-linked diseases, this condition also creates significant medical problems for females carrying only 1 copy of the defective GLA gene. These women may experience many classic symptoms of the disorder including cardiac and kidney problems. However, a small number of females carrying only one copy of the mutated GLA gene never shows any symptoms of Fabry disease.
Cause
Mutations to the GLA gene encoding α-GAL may result in complete loss of function of the enzyme. α-GAL is a lysosomal protein responsible for breaking down globotriaosylceramide, a fatty substance stored various types of cardiac and renal cells.[8] When globotriaosylceramide is not properly catabolized, it is accumulated in cells lining blood vessels in the skin, cells in the kidney, heart and nervous system. As a result, signs and symptoms of Fabry disease begin to manifest.[7]
Treatment
There are two treatment options for Fabry disease: recombinant enzyme replacement therapy and pharmacological chaperone therapy.
Recombinant enzyme replacement therapy (RERT)
RERT was approved as a treatment for Fabry disease in the United States in 2003.[9][10][11]
Two recombinant enzyme replacement therapies are available to functionally compensate for alpha-galactosidase deficiency. Agalsidase alpha and beta are both recombinant forms of the human α-galactosidase A enzyme and both have the same amino acid sequence as the native enzyme. Agalsidase alpha and beta differ in the structures of their oligosaccharide side chains.[12]
In Fabry disease patients, 88% percent of patients develop IgG antibodies towards the injected recombinant enzyme, as it is foreign to their immune system. One suggested approach to solving this problem involves converting the paralogous enzyme α-NAGAL (NAGA) into one that has with α-GAL activity. Because patients still have a functional NAGA gene, their immune system will not produce NAGA antibodies.[13]
The pharmaceutical company Shire manufactures agalsidase alfa (INN) under the trade name Replagal as a treatment for Fabry disease,[14] and was granted marketing approval in the EU in 2001.[15] FDA approval was applied for the United States.[16] However, in 2012, Shire withdrew their application for approval in the United States citing that the agency will require additional clinical trials before approval.[17]
Agalsidase beta
The pharmaceutical company Genzyme produces synthetic agalsidase beta (INN) under the trade name Fabrazyme for treatment of Fabry disease. In 2009, contamination at Genzyme’s Allston, Massachusetts plant caused a worldwide shortage of Fabrazyme, and supplies were rationed to patients at one-third the recommended dose. Some patients have petitioned to break the company’s patent on the drug under the “march-in” provisions of the Bayh–Dole Act.[16]
Pharmacological chaperone therapy
Chaperone mode of action[18][19]
{kind=link}
Fabry patients who display neurological symptoms cannot receive RERT because recombinant enzymes cannot normally pass the blood-brain barrier. Thus, a more suitable alternative treatment is used: pharmacological chaperone therapy.
It has been shown that more potent competitive inhibitors of an enzyme can act as a more powerful chemical chaperone for the corresponding mutant enzyme that fails to maintain proper folding and conformation, despite its intact active site. These chemical chaperones bind to the active site of the mutant enzyme, which can help promote proper folding and stabilize the mutant enzyme. Thus, this results in functional mutant enzymes that will not be degraded via the ubiquitin-proteasome pathway.
1-Deoxygalactonojirimycin (DGJ) has been shown to be both a potent competitive inhibitor of α-GAL and an effective chaperone to for Fabry disease, increasing intracellular α-GAL’s activity by 14-fold.[20][21]
Modifying blood type group B to group O
α-GAL, known as B-zyme in this context, has also demonstrated its ability to convert human blood group B to human blood group O, which can be transfused to patients of all blood types in the ABO blood group categorization. The current B-zyme used comes from Bacteroides fragilis.[19] The idea of maintaining a blood supply at healthcare facilities with all non-O units converted to O units is achieved using enzyme-converted to group O technology, first developed in 1982.[22]
Advantages
A blood bank with ECO blood demonstrates the following advantages:[23]
- Compatible with and transfusable to patients of all blood groups
- Reduce the demand for specific ABO blood groups A, B, AB
- Reduce cost of maintaining a blood bank inventory in hospitals
- Reduce blood transfusion reactions due to human error and ABO incompatibility
- Reduce wastage of less needed blood types
Mechanism of Action
Enzyme converted to type O (ECO) technology to convert blood type B to blood type O.
{kind=link}
Red blood cell (RBC) surfaces are decorated with the glycoproteins and glycolipids that have the same basic sequence with terminal sugar α1‐2‐linked fucose linked to the penultimate galactose. This galactose molecule is called the H antigen.[24][25][26] Blood type A, B, AB, and O differ only in the sugar (red molecule in the illustration) linked with the penultimate galactose. For blood type B, this linked sugar is an α-1‐3‐linked galactose. Using α-GAL, this terminal galactose molecule can be removed, converting RBC to type O.
Supplements
α-GAL derived from aspergillus niger (a common mold) is an active ingredient in products marketed to reduce stomach gas production after eating foods known to cause gas. It is optimally active at 55 degrees C, after which its half-life is 120 minutes.[27]
There are scores of supplements containing the enzyme over the counter in the United States and many more world wide. Products with alpha-galactosidase include:
- Beano
- CVS BeanAid
- Enzymedica’s BeanAssist
- Gasfix
- Bloateez (in India as Cogentrix)
See also
- Beta-galactosidase
- Migalastat, a drug targeting alpha-galactosidase
- Classification of α-galactosidases (according to CAZy)